Pharmaceutical Packaging
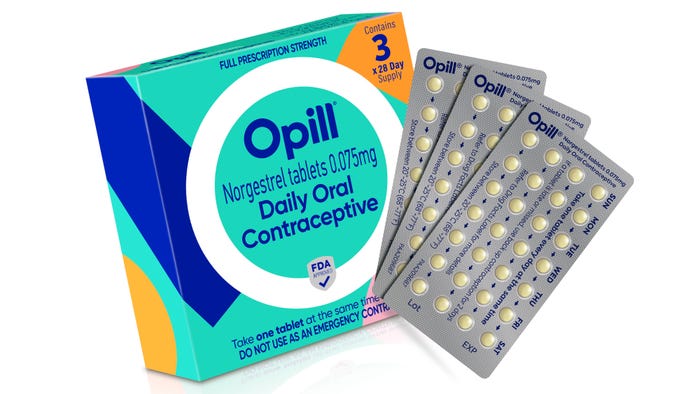
Mylan Levothyroxine 137 mcg
Pharmaceutical PackagingFancy Pharmacy Packs Will Pay DividendsFancy Pharmacy Packs Will Pay Dividends
My recent experience with a prescription refill blew me away. And now I’m going to expect all packs to live up to this high standard.






































Editors' Choice



Sign up for the Packaging Digest News & Insights newsletter.