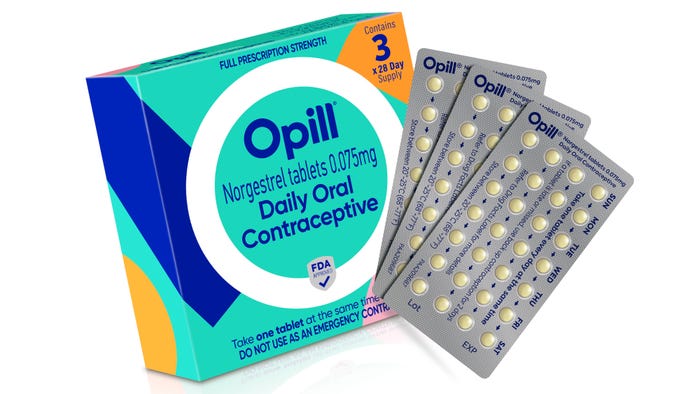
Pharmaceutical Packaging
Thermoform packaging market graphic
Packaging DesignMarket Dynamics Favor Thermoform PackagingMarket Dynamics Favor Thermoform Packaging
Healthy demand from food and pharmaceutical companies will spur growth, despite sustainability challenges.




































Editors' Choice



Sign up for the Packaging Digest News & Insights newsletter.